
February 24th 2021
Marina Emborg, MD PhD, Jeremy Bailoo, PhD and Doris Doudet, PhD
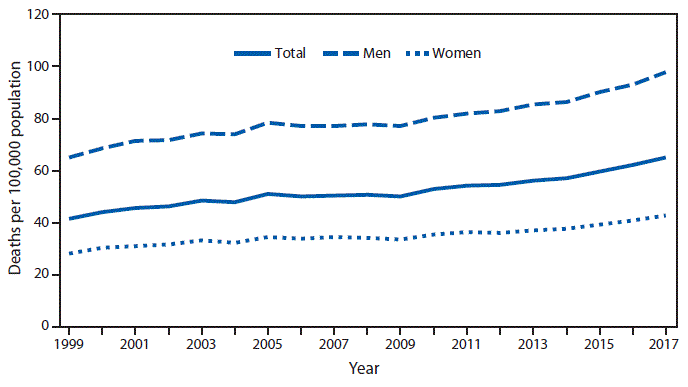
Parkinson’s disease (PD) is the second most common neurodegenerative disease after Alzheimer’s disease. The population prevalence of PD increases from about 1% at age 60 to 4% by age 80. From 1999 to 2017, the age-adjusted death rate for Parkinson disease among adults aged ≥65 years has increased from 41.7 to 65.3 per 100,000 population. PD is more prevalent in men than women.
Typical signs and symptoms of PD include motor disturbances like bradykinesia (slowness of movement), rigidity, postural instability and, in a majority of patients, the characteristic pill rolling tremor. The characteristic underlying pathology of PD motor symptoms is the death of dopamine-generating cells in the substantia nigra, a part of the brain involved in movement, reward and addiction. Non-motor symptoms like constipation, mood and sleep problems often occur before the motor symptoms, and cognitive decline and lower quality of life are common at later stages. Like other common diseases, PD is thought to arise from complex interactions between genetic and environmental factors, which remain mostly unknown.
The history of animal models and Parkinson’s
Animal models have and continue to be important for basic and applied research of human diseases. They are not only important for understanding basic processes underlying disease, but also for the development of disease-specific as well as generalized principles for therapeutic strategies. Further, they are essential for pre-clinical safety and efficacy testing of drugs developed using such research, prior to human clinical trials.
Parkinson’s disease (PD) is not known to spontaneously appear in animals other than humans—in humans the most prevalent presentation of PD is idiopathic (spontaneous) in nature. The features characteristic of PD, however, can be induced through the administration of neurotoxic agents. Using these techniques, the motor symptoms and underlying pathological changes of PD have been researched in drosophila, goldfish, mice, rats, cats, minipigs, sheep, and Central and South American World and Asian and African monkeys.
Most animal models of complex diseases characterize particular aspects of a disease rather than its entirety. The simple reason for this is because most diseases present as a mix of symptoms. Correspondingly, different symptoms may require particular drugs or interventions for treatment. Thus, animal models, much like any model, represent very specific aspects (e.g., symptoms) of the disease it is modeling. If this were not the case, then by definition alone, it could not be termed a model. There are specific standards for a relevant model to meet to be termed as such, including homology of at least some symptoms (face validity), behavioral persistence, similarity to some of the human pathology (construct validity), and improvement in response to effective human therapies (predictive validity)—to name a few. A good model will meet at least some of these criteria. Replicability of the model is also critical so the results can be validated across laboratories. The choice of which symptom to model—the specificity of the model if you will—is prompted by what is known via hypothesis driven research using the scientific method, prioritizing what patients need most.
When considering animal models of PD induced symptoms, there is a long history of animal research which has directly resulted in the therapeutic strategies used to treat humans—a fact which directly refutes claims that induced animal models of PD are of limited value.
Take for example, levodopa (L-DOPA), the most common drug used to control symptoms of PD and Parkinsonism—any condition that causes a combination of the movement abnormalities seen in PD. The use of L-DOPA in alleviating PD related symptoms was discovered by an animal researcher named Arvid Carlsson. He found that rats slowly given reserpine (a drug that depletes dopamine in the brain) lost their ability to move their muscles, and that this state could be rescued by administering L-DOPA. L-DOPA works because it can get inside the brain (cross the blood brain barrier), where neurons use it to make extra dopamine, the missing chemical messenger in PD.

Carlsson’s research launched a prolific body of animal research in the sixties and seventies, where, using rodents and primates, it was discovered that reserpine resulted in the initial release of many neurotransmitters—not just dopamine, followed by the inability to store and depletion of existing stores of neurotransmitters. This knowledge prompted the development of other animal models of induced PD. Among those, the use of toxins like 6OHDA, MPTP or rotenone produced models with various courses and symptoms severity but common motor symptoms of bradykinesia and rigidity. The development of these models also provided clues on mechanisms of neuronal cell loss in PD and the relationship between the exposure to pesticides and increased risk of developing PD. Further animal research demonstrated that prolonged use of L-DOPA in these characterized models results in debilitating involuntary movement (dyskinesia)—a major drug-induced adverse event of PD—thus limiting its use for very long term treatment.
In most cases of PD, however, early L-DOPA treatment of PD guarantees a quality of life standard for many years that could not previously have been realized—a significant fact for those with the disease as well as their caregivers.
That research, in turn, led to the recognition that other drugs derivatives such as amphetamine, MK-801, amantadine and memantine, drugs that increase DA directly or indirectly, were effective in ameliorating PD related symptoms—all made possible thanks to #AnimalResearch. Carlsson subsequently won the Nobel Prize in 2000 in Medicine/Physiology for this research.
Another primary PD treatment with high efficacy is Deep Brain Stimulation (DBS), an invasive surgical procedure which is usually recommended when the L-DOPA-induced dyskinesias become themselves disabling and justify the surgical risk. Notably, DBS was extended to treat many other diseases including Dystonia, Epilepsy, Essential tremor, and obsessive-compulsive disorder (OCD), and is being tested as a treatment for Addiction, Chronic pain, Cluster headache, Dementia, Major Depression, Huntington’s disease, Multiple sclerosis, Stroke recovery, Tourette syndrome and Traumatic brain injury. DBS was logically designed and strictly based on extensive research in the 70s and 80s on the anatomy and normal and pathologic functions of the basal ganglia nuclei. Many open questions about mechanism, efficacy and efficiency, identification of novel brain targets and optimization of current ones have been researched using rats, mice and primates.
The early research using animal models for DBS, however, owes much to basic primate research. Starting in the 1970s, Mahlon DeLong used recordings of individual neurons in various regions of the brain that are part of the basal ganglia, to meticulously characterize the functions of neurons as animals performed movements (e.g., here, here, here and here). Over the next several years, primate research yielded a working understanding of the circuitry underlying motor control in the brain. By this time, researchers understood that it was the loss of dopamine-producing neurons in the basal ganglia neural network that contributed to PD onset and had access to a reliable primate model to study the pathology associated with the symptoms.

In the early 1980s, after reports described how a few young people, in their 20s, developed severe PD-like symptoms after injecting themselves with a synthetic heroin, scientists determined that the cause was an impurity later known as 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine (MPTP). MPTP selectively kills some of the same neurons lost in PD. After it was found that the toxin was ineffective in inducing PD-like motor symptoms in rats but from clinical experience, was highly effective in humans, researchers developed a MPTP-induced non-human primate (monkey) model of PD. The monkey model recapitulated most of the motor symptoms, bradykinesia, rigidity, postural imbalance, freezing but less so of tremor (not a basal ganglia symptom).
DeLong and others used this model to understand how the lack of dopamine leads to motor symptoms of PD and map the circuitry and abnormal firing of its various components. Guided by the circuitry map he previously developed in primates, DeLong lesioned the subthalamic nucleus (STN) in MPTP-treated monkeys and achieved dramatic results: a rapid reduction in PD-like motor symptoms. Studies by other researchers produced similar results, using either surgical lesions or electrodes to induce high frequency stimulation in the STN, internal globus pallidus (GPi) or thalamus. Together, the findings altered prevailing assumptions about how circuit abnormalities might drive motor symptoms, and importantly, provided scientific rationale for targeted therapy.
Meanwhile, with the new understanding of the limitations of L-DOPA treatment for PD, the rapid advancement of medical device technologies, and the new found knowledge of subcortical circuitry, Alim Louis Benabid, a French-Algerian neurosurgeon, proposed in the late 1980s, the development of a device to chronically stimulate the deep nuclei involved in motor control, to reversibly mimic the effects of lesions. Small scale clinical trials in humans with an implantable DBS device developed by Medtronic, a specialized biotech company, which stimulated the ventral intermediate nucleus of the thalamus (VIM) caused PD patients’ tremors to cease. In 1997, the FDA granted approval for Medtronic’s DBS device for VIM-DBS to treat essential tremor and tremor associated with PD.
While DBS in the thalamus reduced tremor, it did not reverse other symptoms reported by patients as even more debilitating, including rigidity and slowed (bradykinesia) or reduced (hypokinesia) voluntary movement, nor did it treat non-motor symptoms that also impact quality of life. Researchers applied DBS to the STN or the GPi in patients with PD. Both improved rigidity and bradykinesia, yielding their best results in patients yet, and successfully bridging basic and clinical neuroscience research. Based on results from a large clinical trial, the FDA approved DBS in the STN/GPI to treat motor symptoms in advanced PD in 2002.
The future of PD research requires primates
Through clinical research, a number of genetic mutations have been found in families of PD patients. Depending on the type of mutation and the population, they can be present in 5% or up to 40% of PD patients. This new knowledge combined with new tools is helping create animal models to study the mechanism that causes neuronal cell death in PD.
The identification of genetic mutations associated with human PD are giving investigators clues about which proteins play a role in neurodegeneration, i.e., how and why neurons die in PD. One of the villains seems to be the neuronal protein α-synuclein, which in PD accumulates and binds to other proteins to form Lewy bodies. A new generation of animal models are being created to study how this protein needed for brain function, over time becomes a neuron “assassin” and to test new therapies. To make this kind of model, investigators may inject directly into the brain, either the faulty protein or a modified virus to produce the protein in excess. Novel technologies to modify genes are being used to introduce the mutations. In addition to anatomical and functional differences between mice, rats, monkeys and humans, genetic differences are also considered for models of genetic PD.
Thus, a primate model is essential for further empirical investigation and progress in our understanding and treatment of PD.
An exciting opportunity of having genetic models of PD is that investigators can study when the disease starts and test for therapies that can prevent neuronal loss, effectively stopping PD in its tracks. That research requires animal research occurring over many years, in the same animals. The availability of non-invasive neuroimaging methods like Magnetic Resonance Imaging (MRI) or Positron Emission Tomography (PET) for large (monkeys) and small animals (rodents) also allows researchers to probe the functional and neurochemical changes during disease progression across time. While some studies can be done in PD patients, long term follow up from initiation, to symptoms progression vs changes in brain chemistry and response to therapies can be assessed in a non-human primate model non-invasively. As the same animals can be studied overtime, fewer animals are used compared to what was needed 3-4 decades ago.
To date, however, there is a need to further develop and refine the models in primates before programmatic research can begin in earnest. Plus, novel therapies for motor and nonmotor symptoms, including methods to replace neurons lost to PD are being created.
This means that we need more, not less primate research. Those opposed to animal research will always do so. For those of us who are considerate of improving the human condition—while weighing the risks and the benefits of doing versus not doing such research—the answer is clearly in favor of animal research for PD in the hopes of finding a cure for this debilitating disease.
